Amide Cyclization is an important way to form peptide cyclization and helix. Depending on its functional groups, a peptide can be cyclized in four different ways: head-to-tail (C-terminus to N-terminus), head-to-side chain, side-chain-to-side-chain, or side chain-to-tail. With our extensive experience in synthesizing cyclic peptides, HongTide can offer high quality amide cyclization peptides.
Head-to-Tail Amide Cyclization
In order to form head-to-tail aminde cyclization, we use 2-CTC-resin. Protect the a-amino group using TFA, DDE group etc. and make the cleavage using low concentration TFA, we can manufacture high quality head-to-tail aminde cyclization peptide with high success rate.
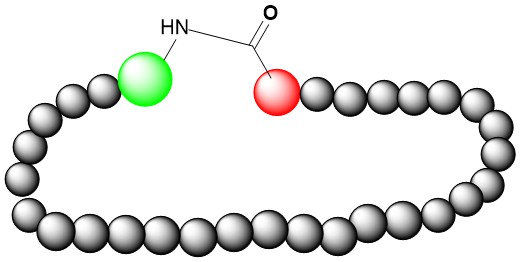
Side Chain-to-Tail Amide Cyclization
Side chain-to-tail amide cyclization can be formed between side chain amide from Lys or Orn and C-terminal carboxyl group if these amino acids exist in peptide sequence. Using 2-CTC-resin, protecting the side chain amino group using TFA, DDE group etc. and making the cleavage using low concentration TFA, we can manufacture high quality Side chain-to-tail amide cyclization peptide with high success rate.
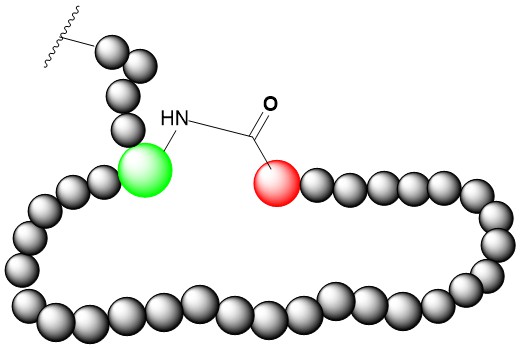
Side Chain-to-Head Amide Cyclization
Side chain-to-head amide cyclization can be formed between side chain carboxyl group from Asp or Glu and N-terminal amide group if these amino acids exist in peptide sequence. Using 2-CTC-resin, protecting the side chain carboxyl group using odmab and de-protect odmab specifically, then making the cleavage using low concentration TFA, we can manufacture high quality Side chain-to-head amide cyclization peptide with high success rate.

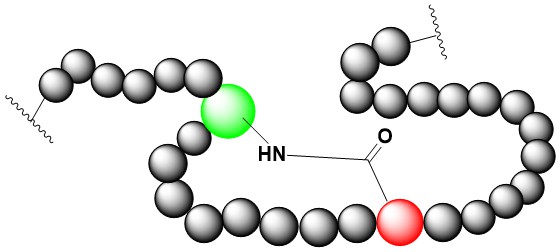
Side-Chain-to-Side-Chain Amide Cyclization
Side-chain-to-side-chain aminde cyclization requires that there are at least one Lys or Orn and one Glu or Asp in the sequence. We can make the side-chain-to-side-chain aminde cyclization before cleavage and after cleavage.
Supplementary Reading: Macrocyclization
Macrocyclization is developed to improve peptide stablity. In all types of cellular processing, protein-protein and protein-peptide interactions play critical roles. Due to their capacity to adapt to the often flexible protein surface, peptides are natural partners of proteins and as ligands, bind to proteins with high affinity. While peptides offer biocompatibility due to their similarity to proteins, they suffer drawbacks that include low plasma bioavailability, instability from proteolytic enzymes, and poor passive membrane permeability as drug candidates. Some success has been achieved with linear peptides, particularly with peptides that maintain α-helical secondary structures. Common peptide-stapling approaches can be introduced to stabilized α-helical motifs, but stapled peptides can suffer from low bioactivity and poor solubility. Another strategy to improve peptide stablity has been to modify peptides by macrocyclization.
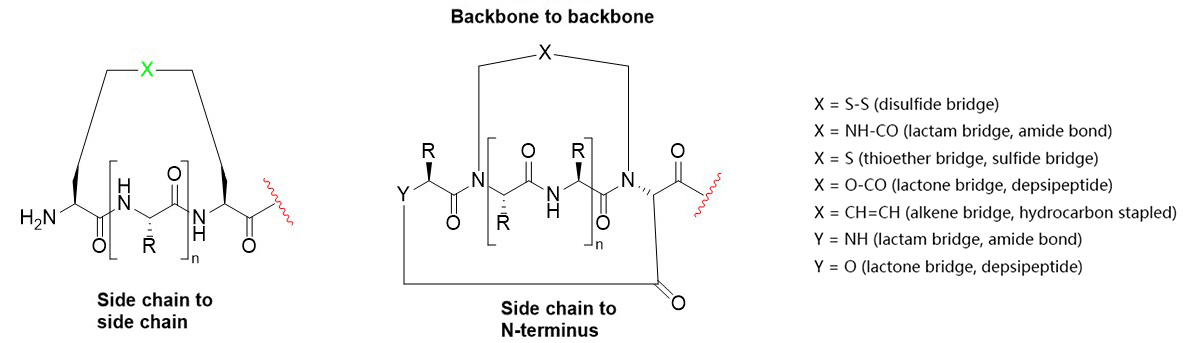
HongTide specializes in the synthesis of complex peptide macrocycles and employs a variety of ring-closure methodologies. We routinely synthesize peptide macrocycles with the following bond types:
-
Multiple, site-selective disulfide bridges (Cys-Cys, Pen-Cys, and Pen-Pen);
-
Amide bond cyclizations (lactamization);
-
Head-to-tail, head-to-side chain, side chain-to-tail, side chain-to-side chain;
-
Backbone-to-backbone, backbone-to-sidechain, backbone-to-head, and backbone-to-tail;
-
Thioether bridges;
-
Hydrocarbon-stapled peptides;
-
Copper-catalyzed azide-alkyne cycloaddition (Click Chemistry).
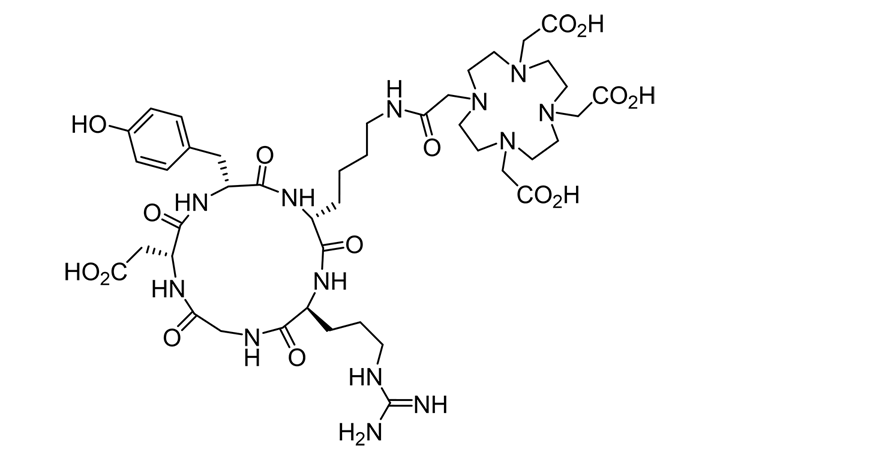
Supplementary Reading: Cyclized RGD
As research and investigation into drug delivery continues, more attention is being placed on cellular uptake and localization. Cell-targeting peptides (CTPs) have developed to target cancer cells that over-express certain receptor proteins, which recognize and internalize CTPs by receptor-mediated endocytosis. Integrins are an important class of heterodimeric transmembrane proteins that play key roles in apoptosis, cell signaling, and cell adhesion. αvβ3 integrins are a best-studied member of the integrin family for their importance in tumor angiogenesis and metastasis. Because αvβ3 integrins are over-expressed in various types of tumor cells (e.g., breast, prostate, and ovarian cancers), and absent in healthy tissue types, they have become an attractive target for cancer therapeutics. Inhibition of αvβ3 integrin receptors has been associated with tumor prevention and reduced tumor growth by antagonizing angiogenesis. Peptide-based antagonists that bind to αvβ3 integrins
have been developed and synthesized. One of the most potent and selective of these peptide antagonists is cyclo[Arg-Gly-Asp-D-Phe-Val] (c[RGDfV]), which was developed by Kessler and co-workers.
Supplementary Reading: RGD-Based Nanomedicines
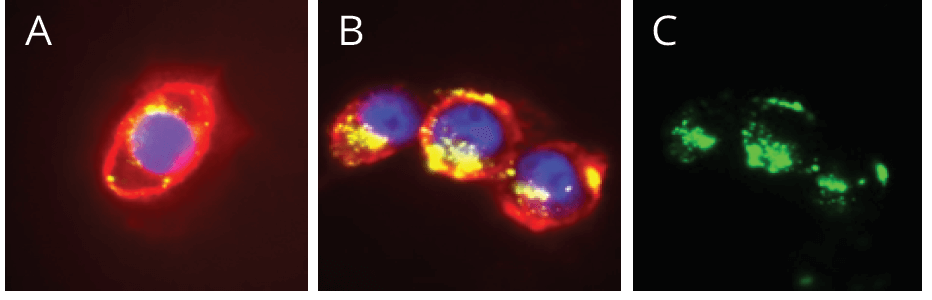
Cyclic RGD (cRGD) peptide-based nanomedicines have been developed for clinical use. Duel targeting liposomal systems that consist of cRGD and transferrin (TF) combined with a liposome (cRGD/ TF-LP) have established a brain glioma cascade delivery system. Crucial to this system was the discovery that cRGD peptide combined with TF enables delivery across the blood-brain barrier (BBB), allowing RGD-targeting in the brain. When combined with paclitaxel, cRGD/TF-LP forms a new system that can precisely target gliomas in the brain, a difficult area for chemotherapy medication alone to reach. Another nanoparticle (NP)-based cRGD targeting system is mesoporous silica nanoparticles (MSNs), a platform that has predominantly been investigated for controlled drug release. MSN-cRGD loaded with camptothecin (CPT) have been used successfuly to target and cause apoptosis in metastatic breast cancer cell lines, MDA-MB 435. By combining the MSN-cRGD platform with a fluorescent tag, it was demonstrated that increase localization and cellular uptake was ocurring in this cell line.
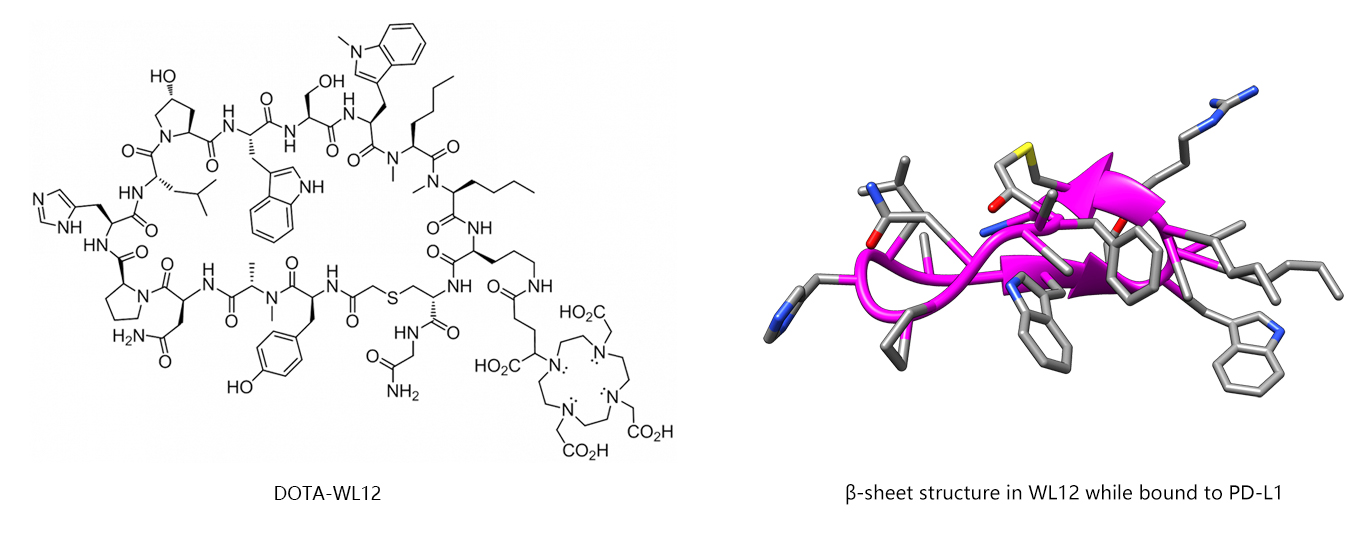
Supplementary Reading: PD-L1 Binding Peptide (WL12)
WL12 is a novel peptide-based PDL1 ligand with a highly specific positron emission tomography (PET) imaging agent. [64Cu]WL12 can be used to detect PD-L1 expression in tumor cells within 60 minutes of radio-tracer administration. The WL12 peptide contains several structural features that make it more resistant to
proteolytic metabolism that include (1) macrocyclization (thioether linkage), (2) main-chain N-methylation, and (3) the incorporation of unnatural amino acids. A DOTA chelator for radiolabeling with 64Cu may be conveniently attached to the primary amine on the ornithine side chain. Modeling studies have suggested that the ornithine residue is not involved in binding to PD-L1. The binding mode of WL12 with PD-L1 involves an intermolecular β-sheet in WL12 that further stabilizes the binding conformation (see below). Stabilization is gained by the addition of two hydrogen bonds formed by an antiparallel edge-to-edge interaction between two opposing strands of the macrocycle. In addition to the β-sheet stabilization, binding is also enhanced by a hydrophobic interaction between D-leucine in WL12 and Ile134 in PD-L1, in which the D-leucine side chain extends into a hydrophobic pocket of PD-L1.
Features














 Contact us by We-chat.
Contact us by We-chat.



